Preamble
- ESPript can either be executed online through a Web interface or at the command-line on Linux operating system.
- The Web version is referred as webESPript in this User Guide.
- The command-line ESPript 3.x binary is freely downloadable (only available for x86-64 Linux OS) - See the F.A.Q. section.
- All commands described below are accessible on webESPript in the EXP MODE .
- Less features are accessible on webESPript in BEG and ADV MODES .
What does the ESPript input file look like in the standalone program?
Line 1: Aligned Sequences
| Content |
Sequence-File |
Selected-Range |
Start-Index |
Extra-Input |
PDB-File |
CNS-File |
| Example |
file.aln |
5-50 |
1 |
+ |
file.pdb |
cns.ctct |
| MODE |
BEG |
ADV |
ADV |
ADV |
EXP |
EXP |
- Sequence-File
File name of the aligned sequences - see Appendix 1 for more details.
- Selected-Range [default: whole sequence]
Range of residues to be displayed (for example 5-50).
- Start-Index [default:
1]
Renumbers residues, so that the first displayed sequence starts with the specified Start-Index.
If the first displayed sequence starts by ATREYES, the command line file.aln 5-4500 2 gives YES and Y is numbered as second residue. Do not Enter a Start-Index value if the first residue is already numbered in file.aln, as explained in Appendix 1. You can check residue numbering of all sequences using option N described in section Output Layout.
- Extra-Input [default: none]
A + specified enables layers or extra input - see layer example for more details.
- PDB-File [default: none]
Name of a PDB file. A PDB output will be generated with occupancy factors replaced by similarity score per residues - see Appendix 2.
- CNS-File [default: none]
Name of a CNS file containing a list of intermolecular contacts - see Appendix 3.
Line 2: Secondary Structures
| Content |
Sec.Str-File |
Acc-Disp |
Sec.Str-File |
Acc-Disp |
ScoreConfidence |
AutomaticSearch |
| Example |
file1.2st |
A |
file2.2st |
A |
9 |
all |
| MODE |
BEG |
BEG |
ADV |
ADV |
ADV |
ADV |
- Sec.Str-File
Name of the file containing secondary structure information. By default, displayed secondary elements are extracted from the first monomer, but you can select a different chainID with the command 'chain_X' (example: file1.2st chain_B).
Three kinds of layout are used, depending if one or two secondary structure files are supplied:
- 1. If one secondary structure file is supplied (uploaded in the TOP secondary structures box in webESPript ):
Secondary structure elements are displayed at the top of each block of sequences and relative accessibility at the bottom.
- 2. If two secondary structure files are supplied:
- secondary structure elements of the first file (uploaded in the TOP secondary structures box in webESPript ) and corresponding accessibility are displayed at the top of each block.
- secondary structure elements of the second file (uploaded in the BOTTOM secondary structures box in webESPript ) and corresponding accessibility are displayed at the bottom of each block.
- 3a. If
file1.2st is entered as usual and the string none is entered as file2.2st: secondary structure elements and relative accessibility are displayed
at the top of each block - see Example 1.
- 3b. If the string
none is entered as file1.2st and file2.2st is entered in turn: secondary structure elements of the second file and relative accessibility are displayed at the bottom of each block.
By default, file1.2st ( TOP secondary structures in webESPript ) and file2.2st ( BOTTOM secondary structures in webESPript ) refer to the first and the last displayed sequences. This default can be changed by using the Special Character X for the first secondary structure file and Z for the second.
Secondary structure elements can be extracted by reading the alignment file file.aln, if you type the character * instead of file1.2st (check Sec. struct. info from PP/NPS@ option in webESPript ). This * option prevents you from typing file.aln twice and can be used for alignment files from PredictProtein or from NPS@, which contain information on predicted secondary structure elements - see Example 1.
- ScoreConfidence [default:
9]
When the secondary structure file is a PHD file, secondary elements with a reliability equal at least to ScoreConfidence are highlighted. If reliability is below limit, helices appear as small squiggles, β-strands as dotted lines and labels are not written - see Example 1.
- AutomaticSearch [default: none]
ESPript searches in the directory $DSSP_DIR (defined as an environment variable) for files having the same name as aligned sequences. Thus, secondary structure information of each aligned sequence with a known 3D structure can be displayed. This option implies that you have the corresponding DSSP files in $DSSP_DIR.
| Content |
Output-file |
NumberingOption |
SequenceOutput |
| Example |
file.ps |
L or M |
SEQ |
| MODE |
BEG |
BEG |
EXP |
- Output-file
Name of the PostScript output file.
- NumberingOption
By default, α-, 310- and π-helices as well as β-strands are numbered with digits.
With the L option, helices and β-strands are numbered with letters, starting at 'A'. With the M option, helices are numbered with digits and strands with letters.
You can remove all secondary structure labels by using the Special Character command: A S all (button Hide labels in webESPript ) if you want to prepare figures such Example 5.
- SequenceOutput
The SEQ option ( Extract reference sequence in webESPript ) allows to extract a single sequence in a one-letter code, from a multiple alignment file entered as file.aln. By default, this sequence correponds to the first displayed in the ESPript figure and is written in a file named file.seq. The extracted sequence can be used in NPS@ or other servers to perform further queries. The SEQ option can also be used to extract sequence information from a PDB file.
| Content |
SimilarityGlobalScore |
SimilarityDiffScore |
SimilarityType |
Consensus |
| Example |
0.7 |
0.5 |
R, B, P, I or S, M, E |
C |
| MODE |
BEG |
ADV |
BEG |
BEG |
- Check Appendix for a general view on similarity computation and colour scheme.
- SimilarityGlobalScore [default:
0.7]
- If R, B, P or I as SimilarityType: a global score is calculated for all sequences by extracting all possible pairs of residues per columns. If applicable, a second score is calculated within each group of sequences.
- If S, M or E as SimilarityType: a percentage is calculated for each column of residues.
If the score is greater than SimilarityGlobalScore, it will be rendered as coloured characters (red characters on a white background by default and white characters on a red background if residues are strictly conserved in the column) with frames (blue by default). Note that strictly conserved residues are boxed but are not framed, if you enter a SimilarityGlobalScore greater than 1.
- SimilarityDiffScore [default:
0.5]
Applicable if R, B, P or I as SimilarityType: residues which are conserved within a group but not conserved from one group to the other are highlighted (yellow background by default).
- SimilarityType [default:
R]
- If R, B, P or I: a matrix is used to calculate the similarity score. Risler, BLOSUM62, PAM250 and Identity are the four possibilities. We recommend a SimilarityGlobalScore of 0.1-0.2 with B or P matrices and of 0.6-0.7 with R or I matrices.
- If S: a percentage of strictly conserved residues is calculated per columns.
- If M: a percentage of similarity is calculated considering criteria used in MultAlin (IV / LM / FY / NDQEBZ).
- If E: a percentage of equivalent residues is calculated per columns, considering physico-chemical properties: HKR are polar positive, DE are polar negative, STNQ are polar neutral, AVLIM are non polar aliphatic, FYW are non polar aromatic, (PGC).
- Consensus [default: none]
A consensus sequence is generated using criteria from MultAlin: uppercase is identity, lowercase is consensus level > 0.5, ! is anyone of IV, $ is anyone of LM, % is anyone of FY, # is anyone of NDQEBZ. lowercase is consensus level > SimilarityGlobalScore if S, M or E are used as SimilarityType.
| Content |
FontSize |
ColumnNb |
Vgap |
Vshift |
Hshift |
Bshift |
PrinterOpt |
Paper |
AllNumbered |
| Example |
7 |
70 |
6 |
0 |
0 |
0 |
C, T, S, B, F |
P, P3, P0, PU, PX,
L, L3, L0, LU, LX |
N |
| MODE |
BEG |
BEG |
BEG |
BEG |
BEG |
ADV |
BEG |
BEG |
BEG |
- FontSize [default:
7]
Size in points for the Courier font (sequence names and residues).
- ColumnNb [default:
60]
Number of residue columns per line.
- Vgap [default:
6]
Vertical gap between two blocks of sequences. The unit for the distance is the height of a line.
- Vshift [default:
0]
Vertical shift for the whole display. The unit for the distance is the height of a line.
- Hshift [default:
0 - centered]
Horizontal shift for the whole display. The unit for the distance is the width of a residue.
- Bshift [default:
0]
Shift lines below bottom sequence. The unit for the distance is the width of a residue.
- PrinterOpt [default:
C]
C coloured output, T coloured with all letters in bold, S light cyan background, B black & white, a grey scale is used ,F flashy colours, similar residues are written with black bold characters and boxed in yellow.
- Paper [default:
P]
P: Portrait A4, P3: Portrait A3, P0: Portrait A0, PU: Portrait US Letter, PX: Portrait 'Tapestry', L: Landscape A4, L3: Landscape A3, L0: Landscape A0, LU: Landscape US Letter, LX: Landscape 'Tapestry'.
- AllNumbered [default: first sequence]
By default, the first sequence is numbered every ten residues as in Example 2. With the option N (check Number sequences option in webESPript ) all sequences are numbered at the beginning of each block of sequences as in Example 3.
Lines 6: Special Commands
• Hide sequences
Aligned sequences are not written (check Hide sequences in webESPript ). This option can be used to build a figure including several secondary structure elements as in Example 4. @skip is a shortcut for the block of Special Characters below:
I S all ! skip all
F S all ! skip all
H S all ! skip all
B S all ! skip all
O S all ! skip all
N S all ! skip all
T S all ! skip all
Y S all ! skip all
• More info from a file from PredictProtein or NPS@
Extra information can be extracted upon use of the command @pp if:
- A result file from the PredictProtein server is entered as file.aln:
ProDom domains are visualized with yellow bars below each block of sequences. 'x' marks from the SEG low-complexity (1) search are represented with dotted lines. Peptides resulting from a PROSITE (2) search are shown with bold letters.
- A file from the NPS@ server with multiple sequence alignment and predicted secondary structure elements is entered as file.aln:
Predicted secondary structure elements are shown below each aligned sequence (i.e. helices with squiggles, β-strands with arrows, ambiguous predictions with solid circles).
• Minus / Plus
| Example |
@minus 5 40
@plus 63 |
| MODE |
EXP |
Residue numbering can be changed along a single sequence.
If @minus is used, numbering is shifted by -1 at the given column (here at columns 5 and 40). If @plus is used, residue numbering is shifted by +1 at the given column. Before using this option, use the command @ruler described below to visualize column numbers.
@minus and @plus are equivalent to the options Delete in seq numbering and Insert in seq numbering in webESPript .
Note that by default sequence numbering refers to the first displayed sequence, but it can refer to the third displayed sequence (for example) if you enter the Special Command Y D 3.
• Preview column numbers
Column numbers are displayed. This option is useful when preparing a figure with the special commands @minus or @plus presented above, or the Special Characters Q, V, W.
• Insert text at sequences
| Example |
@seq 5 text
@seq vp7_ehdv1 text |
| MODE |
EXP |
The command is: @seq [sequence number or sequence name] [text or blank]
The text is then inserted above the chosen sequence. Note that sequences numbers are given in the log file of ESPript.
Special case: the text is inserted below the last displayed sequence, if you chose a number greater than the number of displayed sequences. Thus, you can give a name to a line of Special Characters and change the colour of the name with the Special Character T.
• Modify or create colours
| Example |
@col R .8 0 0
@col B 0 0 .8 |
| MODE |
EXP |
Assigns a new RGB code for a Special Characters colour in ESPript. You can also create a new special character colour, such as A for grey:
@col A .5 .5 .5 ! create a new colour named A
I A all ! strictly conserved residues are in grey
Remark: a new character colour must be created before being used as in the example above. S is reserved to skip. Otherwise, any uppercase character can be used. Have a look at this site to chose new colours and corresponding percent RGB values (range is 0.0-1.0 and white is 1 1 1).
• Replace labels
| Example |
@aA1 aA2 bB1 hH1 bB2
@aA3 bB3 |
| MODE |
ADV |
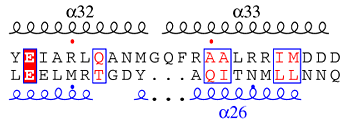
Secondary structure labels can be replaced by new ones defined by the user. Labels starting by a, b, h, p refer to α-helices, β-strands, 310-helices and π-helices respectively. These first characters are not displayed. Replacement is made according to the order of entrance (see Example 4), firstly through the top secondary structure elements, then through the bottom secondary structure elements, if applicable.
Command lines can be written with all α-helices firstly, then all β-strands, 310- and π-helices. For instance you can remove labels of all 310-helices by typing as many @h h h h h as needed.
If the first letter is typed in uppercase (@Ag1 Ag2), the second letter is displayed using a Symbol font (here, displayed labels would be γ1 γ2).
• Hide turns
Strict α- and β-turns, usually rendered as TTT and TT, are not displayed (see information on secondary structures).
• Insert secondary structure elements
| Example |
@top a 10-20 20-30 b 50-55
@bottom b 25-35 |
| MODE |
EXP |
Inserts α-helices (a), β-strands (b), 310-helices (h) or π-helices (p) at the top or bottom of sequences blocks. Rules of numbering are the same as in section Secondary Structures (i.e. by default, top and bottom secondary structure elements match top and bottom sequences, respectively).
You can enter up to 264 characters on this line of command. Click on the button +1 of the interface to duplicate the form if you exceed this limit. Thus, you may be able to enter α-helices in Layer 0 and β-strands in Layer 1 , while still being under the limit of 264 characters in each part.
• Hide names of secondary structure elements
Removes the name of the corresponding sequence at the beginning of each line of secondary structure elements. By default, this name has the same colour as the first displayed element.
Remark: assume a very special case, where your sequence starts at 10, and you want to colour secondary structure name in red and secondary structure elements in blue. Then you can use the Special Characters command X:
X R 10-10
X B 11-4500
• Hide alternate conformations
Removes grey stars added on the top of blocks of sequences, above residues with alternate conformations.
• Hide disulphide bridges
Removes green digits (1 1, 2 2...) added on the figure at the bottom of sequences blocks to show disulphide bridges.
• Substitute sequence names
| Example |
@sub oldname1 newname1 oldname2 newname2 |
| MODE |
EXP |
Replaces the name of a sequence contained in your alignment file file.aln by a new one. You can substitute up to 15 names.
Suppose you want to change the names of the first and third displayed sequences, you can enter: @sub 1 newname1 3 newname2
• Color by residues physicochemical properties
The residues are coloured according to their physico-chemical properties.
Lines 6b: Special Characters
| Content |
Character-Type |
Character-Colour |
Position |
| Options |
P, T, R, X, Y, Z, Q, V, W, U, D, G, J, S, C, E, L, K, A, I, F, M, H, B, O, N, s, t, u, a, b, c, d, e, f, g, h, i, j, k, l, m, n |
D, B, R, P, G, F, C, O, Y, M, W, S |
X Y-Z |
| Example |
U R 2 9-39 |
| MODE |
ADV |
Entry on each line is: Character-Type Colour Position
example: U R 2 9-39 adds red (R) triangles (U) at residue 2 and at residues 9 to 39 (2 9-39)
Character-Type
|
|
Miscellaneous
|
P | calculates hydropathy |
T | changes colour of sequence names |
R | reads intermolecular contacts |
|
Assignment
|
X | top secondary structure information is assigned to a chosen sequence, which is the first one by default. Colour of secondary elements can be changed. |
Y | sequence numbering is assigned to a chosen sequence, which is the first one by default. Colour of digits can be changed. |
Z | residue numbering of another sequence, which is the last one by default, can be displayed at the bottom of sequences blocks. Secondary structure information corresponding to this sequence can also be displayed (see Example 3). |
|
Do it yourself
|
Q | boxes residues (see Example 5) |
V | bold characters |
W | adds frames |
|
Changing default colours of
|
A | labels above top secondary structure elements |
I | identity boxes |
F | identity characters |
M | group similarity boxes |
H | group similarity characters |
B | global similarity frames |
O | difference similarity boxes |
N | low similarity scores |
|
Adding markers
|
U | triangle up (see Example 2) |
D | triangle down |
G | go |
J | jammed |
S | star |
C | solid circle |
E | open circle |
L | dotted line |
K | stroke |
|
Adding NMR markers
|
s | amide proton slow exchange rate (< 1mn-1) |
t | 3JHN,Hα NH-Hα coupling constant < 6 Hz |
u | 3JHN,Hα NH-Hα coupling constant ≥ 7 Hz |
a, b, c | dNN(i,i+1) NOE between proton NH of residue i and i+1 (weak, medium, strong) |
d, e, f | dαN(i,i+1) NOE between proton α of residue i and proton NH of i+1 (weak, medium, strong) |
g, h, i | dβN(i,i+1) NOE between proton β of residue i and proton NH of i+1 (weak, medium, strong) |
j | dNN(i,i+2) NOE between proton NH of residue i and proton NH of i+2 |
k | dαN(i,i+2) NOE between proton α of residue i and proton NH of i+2 |
l | dαN(i,i+3) NOE between proton α of residue i and proton NH of i+3 |
m | dαβ(i,i+3) NOE between proton α of residue i and proton β of i+3 |
n | dαN(i,i+4) NOE between proton α of residue i and proton NH of i+4 |
Character-Colour
(except if R is Character-Type)
|
D
Black |
B
Blue |
R
Red |
P
Pink |
G
Green |
F
Green fluo |
C
Cyan |
O
Orange |
Y
Yellow |
M
Maroon |
W
White |
S
Transparent |
Position
By default, residues are numbered according to the first displayed sequence
[ ] means mandatory and { } optional
|
| 1 |
if Character-Type= P, T
[sequence name number or range] {other sequence name number or range} {...}
|
Example 1: to calculate hydropathy of the third displayed sequence: P R 3 (the string hyd will be written in red)
Example 2: to colour the name of the second sequence in green: T G 2
|
| 2 |
if Character-Type= R
[chainId] [residue range] {other residue range} {...}
|
See Appendix for details on intermolecular contacts
|
| 3 |
if Character-Type= X, Y, Z
[name or number of sequence displayed] {Start-Index (1 by default)}
or
[residue range] {other residue range} {...}
|
Example 1: to assign the first secondary structure file to the third displayed sequence: X B 3 (sec. structure elements are in blue)
Example 2: to number the fourth displayed sequence in blue: Z B 4
(the same command Z B 4 can be used to assign the second sec. structure file to the fourth displayed sequence and to colour sec. structure elements in blue).
Example 3: to colour elements in blue and red:
X B 3 (secondary structure elements refer to the 3 displayed sequence and are in blue. This sequence is now the reference)
X R 4-50 60-80 (but secondary structure elements from residues 4 to 50 and from 60 to 80 are in red)
Remark: you can type X B name_of_the_third_displayed_sequence instead of X B 3
|
| 4 |
if Character-Type= Q, V, W
[number or range of sequence displayed] {column range} {other column range} {...}
|
Note that, here, column numbering is used instead of residue numbering. Use the command @ruler to preview column numbers.
Example 1: to highlight in yellow residues of sequences 3-8 from columns 40 to 45 and from 50 to 55: Q Y 3-8 40-45 50-55
Example 2: to highlight the last sequence in cyan: Q C 1000
|
| 5 |
if Character-Type= U, D, S, C, L, A, I, F, M, H, B, O, N, s, t, u, a, b, c, d, e, f, g, h, i, j, k, l, m, n
[residue number or range] {other residue number or range} {...}
|
Example 1: to add red triangles at residue 2 and from 9 to 39: U R 2 9-39
Example 2: to box all identical residues in blue: I B 1-4500
Example 3: to remove all secondary structure labels: A S 1-4500
By default, positions refer to residue numbering of the first displayed sequence. Use the special command Y to change this default:
Y B 3 (residue numbering refers to the 3 displayed sequence and residues numbering is in blue)
U R 9 20-30 (adds red triangles below columns containing residues 9 and 20 to 30 of sequence 3)
|
| Example |
%This is a reminder |
| MODE |
ADV |
A line beginning with % will be displayed at the bottom of the generated PostScript, as a comment or a title.
Line 6d: Ending the section
A single dot on a line ends this section.
Lines 7: Defining Groups and Blocks
| Example |
1-4 9 %8
6 5 7
. |
| MODE |
BEG |
You can select the sequences to be displayed and their order on a single line: 2 1 3-5
all can be used to select the rest of the sequences: 2 all (see Example 5).
A % before a sequence number keeps a sequence for similarity calculations but prevents it from displaying: 2 %1 %3-5 (see Example 4).
You can also separate your sequences in groups for similarity computations, each line defining a group and giving the order of the sequences to display as in Example 2 ( ADV or EXP modes in webESPript ). The calculation by group is not performed if SimilarityType is Strict, Multalin or Equivalent (groups are just numbered).
This section is ended by a single dot on a single line.
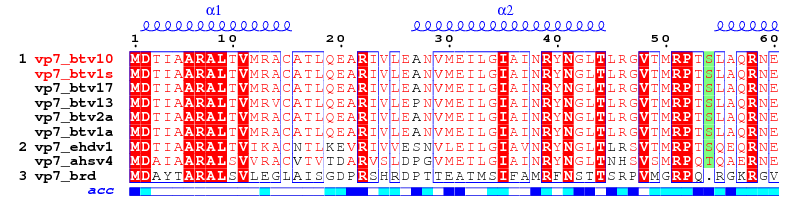
• file.aln
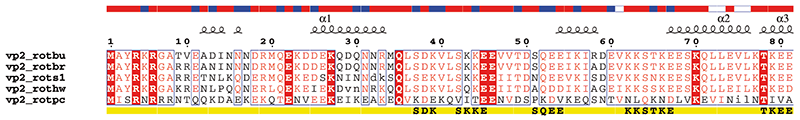
file.aln is an ASCII file containing aligned sequences. The following formats are supported:
Should you have other aligned sequences, be sure to keep two fields per line: the first one is the name of the sequence, the second one the sequence itself. Use white characters (spaces) to separate the two fields; use blank lines to separate two blocks as in:
vp7_btv1s MDTIAARALTVMRACATLQEARIVLEANVMEILGIAINRYNGLTLRGVTMRPTSLAQRNE
vp7_btv10 MDTIAARALTVMRACATLQEARIVLEANVMEILGIAINRYNGLTLRGVTMRPTSLAQRNE
vp7_btv1s MFFMCLDMMLSAAGINVGPISPDYTQHMATIGVLATPEIPFTTEAANEIARVTGETSTWG
vp7_btv10 MFFMCLDMMLSAAGINVGPISPDYTQHMATIGVLATPEIPFTTEAANEIARVTGETSTWG
FASTA format for multiple alignments is supported. Sequences can be entered as below:
> vp7_btv1s
MDTIAARALTVMRACATLQEARIVLEANVMEILGIAINRYNGLTLRGVTMRPTSLAQRNE
MFFMCLDMMLSAAGINVGPISPDYTQHMATIGVLATPEIPFTTEAANEIARVTGETSTWG
> vp7_btv10
MDTIAARALTVMRACATLQEARIVLEANVMEILGIAINRYNGLTLRGVTMRPTSLAQRNE
MFFMCLDMMLSAAGINVGPISPDYTQHMATIGVLATPEIPFTTEAANEIARVTGETSTWG
If a Start-Index is present in file.aln (at least in the first block of sequences), residue numbering is modified accordingly.
Format is title_Start-Index_ or title(Start-Index) as below:
vp7_btv1s(3) TIAARALTVMRACATLQEARIVLEANVMEIL
vp7_btv10(5) --AARALTVMRACATLQEARIVLEANVMEIL
vp7_btv1s GIAINRYNGLTLRGVTMRPTSLAQRNEMFFM
vp7_btv10 GIAINRYNGLTLRGVTMRPTSLAQRNEMFFM
• file.pdb
You can enter the name of a PDB file at the first input line (instead of the multiple alignment file, file.aln). ESPript will extract a one letter code sequence, corresponding to all the residues contained in this PDB file. You can display the sequence of a single monomer defined by a chainID in the PDB file, by using the command chain_X on the input line: file.pdb chain_A
The extracted sequence is given the name of the input PDB file. This default can be changed, if the header of the PDB file contains a line starting by DBREF. The string of characters following DBREF will be the name of the extracted sequence: DBREF sequence_name
You can also enter the name of a multiple alignment file, file.aln, and of a PDB file, file.pdb, on the first input line: file.aln file.pdb (see Example 2).
Then, a file named file_bcol.pdb is created by ESPript from file.pdb. The occupancy factors of the original file file.pdb are replaced by similarity scores in file_bcol.pdb.
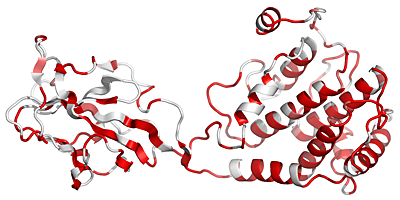
Attention, similarity scores in file_bcol.pdb have been rescaled between 0 and 100. This trick allows in a next step, to show conserved region along the structure with a nice colour ramping going from white to red. The command chain_X allows to copy the similarity score of a chosen monomer in the output file file_bcol.pdb: file.aln file.pdb chain_A
The output PDB file, file_bcol.pdb, is used to produce a PyMOL cartoon representation as shown below (to that end, check Generate a PyMOL view ).
Residues with SimilarityGlobalScore lower than 0.7 are in white, conserved areas with SimilarityGlobalScore in the range 0.7-1.0 are colour-ramped in red with a 0-100 pseudo occupancy factor value.
• Intermolecular contacts
A log file produced by CNS (11) can be read by ESPript to display protein:protein contacts (see Example 4). You can also use ENDscript to generate rapidly such a figure. A list of contacts is generated as follows:
- Crystallographic contacts - addition to CNS command file:
delete selection=(hydrogen) end flags exclude * include pvdw end
parameter nbond wmin=4.0 end end energy end
generates in CNS log file:
%atoms "A -62 -ASN -OD1 " and "C -112 -THR -C "(XSYM# 4) only 3.64 A apart
- Non-crystallographic contacts - addition to CNS command file:
flags exclude * include vdw end parameter nbond wmin=0 end end
distance cuton=0.0 cutoff=4 from =(segid A) to =(not segid A) end
generates in CNS log file:
atoms "A -90 -ALA -CB " and "B -181 -HIS -CE1 " 3.6958 A apart
Residue names, residue numbers, first letter of chainIDs and distances are extracted from the CNS log file. If the input line in ESPript is R A all, chainIDs of all residues in contact
with molecule A are displayed on a bottom line named i_A. The chainID character is in red if the distance is less than 3.2 Å and in black if it is in the range 3.2-4.0 Å. The shortest intermolecular distance is taken for each residue. Thus, a B would be written under residue 90, if the distance listed in the example above is the shortest between Ala90 chainID A and His181 chainID B. A A would be written under His181 on a new bottom line named i_B with the command R B all.
Contacts can be further analysed looking to the figure produced by ESPript:
- A to Z, 0 to 9 or a to z means that the concerned amino acid residue has a non-crystallographic contact with an amino acid residue of the Chain A to Z, 0 to 9 or a to z (e.g. this amino acid residue is involved in a non-crystallographic interface).
- A to Z, 0 to 9, a to z in italic means that the concerned amino acid residue has a crystallographic contact with an amino acid residues of the Chain A to Z, 0 to 9 or a to z (e.g. this amino acid residue is involved in a crystallographic interface).
- # identifies a contact between two amino acid residues having the same names and numbers (e.g. along a 2-fold symmetry axis).
• file.2st
This file is an ASCII file from which ESPript will extract secondary structure information. The following formats are supported:
DSSP (12) (a PDB file can be directly uploaded if you use webESPript , DSSP being executed on the server)
STRIDE (13)
PHD (14)
α-helices, 310-helices and π-helices are displayed as medium, small and large squiggles respectively. β-strands are rendered as arrows, strict β-turns as TT letters and strict α-turns as TTT. The secondary structures files of the two sequences have been entered in the excerpt below.
A verification is performed between residue names of the secondary structure file and of the chosen sequence (which is the first displayed by default). In case of problem, the program will try to align the two sequences without gaps. You get the following warnings, if some residues do not correspond between the two sequences:
Warning: DSSP residue M does not match seq residue D 2 sequence 1 column 2
If the program failed to align the two sequences, you get an error message:
Warning: DSSP residue M does not match seq residue D 2 sequence 1 column 2
Warning: DSSP residue D does not match seq residue T 3 sequence 1 column 3
...........................
Error: sec. structure elements are certainly misplaced
and the figure generated by ESPript gives you a false information.
A file produced by DSSP can include the positions of disulphide bridges. This information is rendered in ESPript by green digits (1 1, 2 2 ...) written under each column with a bound cystein.
Residues with alternate positions can also be flagged in the DSSP file (we use a modified version of DSSP on webESPript ), in order to be marked by grey stars on the top of sequences blocks in the PostScript figure.
• Accessibility
The relative accessibility of each residue can be extracted from DSSP (12) and PHD (14) files. It is rendered as blue-coloured boxes located at the last or first line of each block (see Secondary Structures). Note that DSSP include only protein atoms in its calculation of accessibility. Coordinates of water molecules, ligands... are not taken into account. The blue square scale is set as follow:
| colour |
value |
accessibility |
| blue |
0.4 < A ≤ 1.0 |
accessible |
| cyan |
0.1 ≤ A ≤ 0.4 |
intermediate |
| white |
A < 0.1 |
buried |
| blue with red borders |
A > 1.0 |
|
| red |
either accessibility is not predicted in PHD (14) or residue names between sequence and DSSP (12) file do not match
|
• Hydropathy
The hydropathic character of a sequence selected with the P command (P D 1 for first displayed sequence) is calculated according to the algorithm of Kyte & Doolittle (15) with a window of 3.
| colour |
values |
Hydropathy |
| pink |
H > 1.5 |
hydrophobic |
| grey |
-1.5 ≤ H ≤ 1.5 |
intermediate |
| cyan |
H < -1.5 |
hydrophilic |
| Hydropathic values for each residue |
I |
V |
L |
F |
C |
M |
A |
G |
T |
S |
W |
Y |
P |
H |
E |
Q |
D |
N |
K |
R |
| 4.5 |
4.2 |
3.8 |
2.8 |
2.5 |
1.9 |
1.8 |
-0.4 |
-0.7 |
-0.8 |
-0.9 |
-1.3 |
-1.6 |
-3.2 |
-3.5 |
-3.5 |
-3.5 |
-3.5 |
-3.9 |
-4.5 |
• Similarity scores
If Risler BLOSUM62 PAM250 or Identity , several scores are calculated:
- in-Group Score (ISc) is a classical computation of a similarity score within each group.
For a column made of 3 residues ACD:
ISc = (AC+AD+CD) ÷ 3
- Cross-Group Score (XSc) is the similarity score average for every sequence pair, where each sequence belongs to a different group.
For a column made of 6 residues divided in 3 groups (ACD)(DE)(G):
XSc = [(AD+AE+CD+CE+DD+DE)÷6+(AG+CG+DG)÷3+(DG+EG)÷2] ÷ 3
- Total Score (TSc) is the mean of in-Group Score and Cross-Group Score:
TSc = (ISc + XSc)÷2
The user specifies a threshold for in-Group (ThIn) and Diff-Group (ThDiff) scores.
Colours are chosen according to the following rule:
A
Red box, white character → Strict identity.
Y
Red character (or black bold character with color scheme "Flashy") → Similarity in a group: ISc > ThIn.
T
Blue frame (filled in yellow with color scheme "Flashy") → Similarity across groups: TSc > ThIn.
Q
Green fluo box → Differences between conserved groups: (ISc-Xsc)÷2 > ThDiff.
• Similarity scores matrices
Risler matrix (16)
A C D E F G H I K L M N P Q R S T V W Y .
A 22-15 2 17 6 6 -6 17 14 13 10 13 -2 18 15 20 19 20 -9 2-30
C-15 22-17-15-16-17-18-16-16-15-16-16-18-14-15-13-14-14-18-11-30
D 2-17 22 10 -3 -4-13 0 1 -2 -5 8-12 6 -1 7 0 0-14 -4-30
E 17-15 10 22 6 3 -6 15 14 9 6 14 -1 21 19 18 16 16-10 2-30
F 6-16 -3 6 22 -4-11 10 1 10 -2 4-11 7 4 5 3 8 -9 20-30
G 6-17 -4 3 -4 22-12 0 -1 -2 -4 2-12 2 1 7 2 1-13 -2-30
H -6-18-13 -6-11-12 22 -8-10 -9-12 -3-16 -5 -4 -4 -9 -7-17 -8-30
I 17-16 0 15 10 0 -8 22 10 21 9 9 -6 14 14 16 16 22 -7 4-30
K 14-16 1 14 1 -1-10 10 22 7 4 10 -7 17 21 14 12 12-11 5-30
L 13-15 -2 9 10 -2 -9 21 7 22 18 8 -8 11 12 13 12 20 -8 5-30
M 10-16 -5 6 -2 -4-12 9 4 18 22 0-12 12 11 6 8 8-13 -2-30
N 13-16 8 14 4 2 -3 9 10 8 0 22-10 16 12 19 11 11-11 -1-30
P -2-18-12 -1-11-12-16 -6 -7 -8-12-10 22 -6 -3 -3 -5 -6-16-12-30
Q 18-14 6 21 7 2 -5 14 17 11 12 16 -6 22 20 18 17 15-10 5-30
R 15-15 -1 19 4 1 -4 14 21 12 11 12 -3 20 22 20 19 15 -8 8-30
S 20-13 7 18 5 7 -4 16 14 13 6 19 -3 18 20 22 21 18 -8 4-30
T 19-14 0 16 3 2 -9 16 12 12 8 11 -5 17 19 21 22 16-10 3-30
V 20-14 0 16 8 1 -7 22 12 20 8 11 -6 15 15 18 16 22 -7 3-30
W -9-18-14-10 -9-13-17 -7-11 -8-13-11-16-10 -8 -8-10 -7 22 -6-30
Y 2-11 -4 2 20 -2 -8 4 5 5 -2 -1-12 5 8 4 3 3 -6 22-30
.-30-30-30-30-30-30-30-30-30-30-30-30-30-30-30-30-30-30-30-30 0
|
PAM250 matrix (17)
A R N D C Q E G H I L K M F P S T W Y V .
A 2 -2 0 0 -2 0 0 1 -1 -1 -2 -1 -1 -4 1 1 1 -6 -3 0-15
R -2 6 0 -1 -4 1 -1 -3 2 -2 -3 3 0 -4 0 0 -1 2 -4 -2-15
N 0 0 2 2 -4 1 1 0 2 -2 -3 1 -2 -4 -1 1 0 -4 -2 -2-15
D 0 -1 2 4 -5 2 3 1 1 -2 -4 0 -3 -6 -1 0 0 -7 -4 -2-15
C -2 -4 -4 -5 12 -5 -5 -3 -3 -2 -6 -5 -5 -4 -3 0 -2 -8 0 -2-15
Q 0 1 1 2 -5 4 2 -1 3 -2 -2 1 -1 -5 0 -1 -1 -5 -4 -2-15
E 0 -1 1 3 -5 2 4 0 1 -2 -3 0 -2 -5 -1 0 0 -7 -4 -2-15
G 1 -3 0 1 -3 -1 0 5 -2 -3 -4 -2 -3 -5 -1 1 0 -7 -5 -1-15
H -1 2 2 1 -3 3 1 -2 6 -2 -2 0 -2 -2 0 -1 -1 -3 0 -2-15
I -1 -2 -2 -2 -2 -2 -2 -3 -2 5 2 -2 2 1 -2 -1 0 -5 -1 4-15
L -2 -3 -3 -4 -6 -2 -3 -4 -2 2 6 -3 4 2 -3 -3 -2 -2 -1 2-15
K -1 3 1 0 -5 1 0 -2 0 -2 -3 5 0 -5 -1 0 0 -3 -4 -2-15
M -1 0 -2 -3 -5 -1 -2 -3 -2 2 4 0 6 0 -2 -2 -1 -4 -2 2-15
F -4 -4 -4 -6 -4 -5 -5 -5 -2 1 2 -5 0 9 -5 -3 -3 0 7 -1-15
P 1 0 -1 -1 -3 0 -1 -1 0 -2 -3 -1 -2 -5 6 1 0 -6 -5 -1-15
S 1 0 1 0 0 -1 0 1 -1 -1 -3 0 -2 -3 1 2 1 -2 -3 -1-15
T 1 -1 0 0 -2 -1 0 0 -1 0 -2 0 -1 -3 0 1 3 -5 -3 0-15
W -6 2 -4 -7 -8 -5 -7 -7 -3 -5 -2 -3 -4 0 -6 -2 -5 17 0 -6-15
Y -3 -4 -2 -4 0 -4 -4 -5 0 -1 -1 -4 -2 7 -5 -3 -3 0 10 -2-15
V 0 -2 -2 -2 -2 -2 -2 -1 -2 4 2 -2 2 -1 -1 -1 0 -6 -2 4-15
.-15-15-15-15-15-15-15-15-15-15-15-15-15-15-15-15-15-15-15-15 0
|
BLOSUM62 matrix (18)
A R N D C Q E G H I L K M F P S T W Y V .
A 4 -1 -2 -2 0 -1 -1 0 -2 -1 -1 -1 -1 -2 -1 1 0 -3 -2 0 -4
R -1 5 0 -2 -3 1 0 -2 0 -3 -2 2 -1 -3 -2 -1 -1 -3 -2 -3 -4
N -2 0 6 1 -3 0 0 0 1 -3 -3 0 -2 -3 -2 1 0 -4 -2 -3 -4
D -2 -2 1 6 -3 0 2 -1 -1 -3 -4 -1 -3 -3 -1 0 -1 -4 -3 -3 -4
C 0 -3 -3 -3 9 -3 -4 -3 -3 -1 -1 -3 -1 -2 -3 -1 -1 -2 -2 -1 -4
Q -1 1 0 0 -3 5 2 -2 0 -3 -2 1 0 -3 -1 0 -1 -2 -1 -2 -4
E -1 0 0 2 -4 2 5 -2 0 -3 -3 1 -2 -3 -1 0 -1 -3 -2 -2 -4
G 0 -2 0 -1 -3 -2 -2 6 -2 -4 -4 -2 -3 -3 -2 0 -2 -2 -3 -3 -4
H -2 0 1 -1 -3 0 0 -2 8 -3 -3 -1 -2 -1 -2 -1 -2 -2 2 -3 -4
I -1 -3 -3 -3 -1 -3 -3 -4 -3 4 2 -3 1 0 -3 -2 -1 -3 -1 3 -4
L -1 -2 -3 -4 -1 -2 -3 -4 -3 2 4 -2 2 0 -3 -2 -1 -2 -1 1 -4
K -1 2 0 -1 -3 1 1 -2 -1 -3 -2 5 -1 -3 -1 0 -1 -3 -2 -2 -4
M -1 -1 -2 -3 -1 0 -2 -3 -2 1 2 -1 5 0 -2 -1 -1 -1 -1 1 -4
F -2 -3 -3 -3 -2 -3 -3 -3 -1 0 0 -3 0 6 -4 -2 -2 1 3 -1 -4
P -1 -2 -2 -1 -3 -1 -1 -2 -2 -3 -3 -1 -2 -4 7 -1 -1 -4 -3 -2 -4
S 1 -1 1 0 -1 0 0 0 -1 -2 -2 0 -1 -2 -1 4 1 -3 -2 -2 -4
T 0 -1 0 -1 -1 -1 -1 -2 -2 -1 -1 -1 -1 -2 -1 1 5 -2 -2 0 -4
W -3 -3 -4 -4 -2 -2 -3 -2 -2 -3 -2 -3 -1 1 -4 -3 -2 11 2 -3 -4
Y -2 -2 -2 -3 -2 -1 -2 -3 2 -1 -1 -2 -1 3 -3 -2 -2 2 7 -1 -4
V 0 -3 -3 -3 -1 -2 -2 -3 -3 3 1 -2 1 -1 -2 -2 0 -3 -1 4 -4
. -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 -4 1
|
Identity matrix
A R N D C Q E G H I L K M F P S T W Y V .
A 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
R 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
N 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
D 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
C 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
Q 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
E 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0
G 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0
H 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0
I 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0
L 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0 0
K 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0 0
M 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0 0
F 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0 0
P 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0 0
S 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0 0
T 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0 0
W 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0 0
Y 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0 0
V 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 1 0
. 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
|
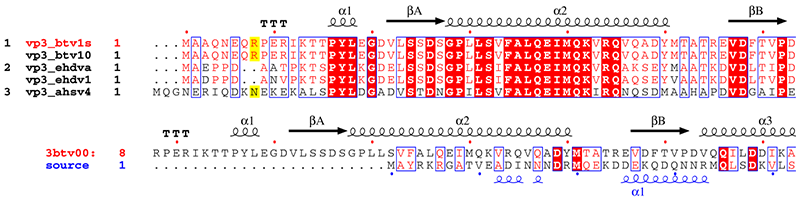
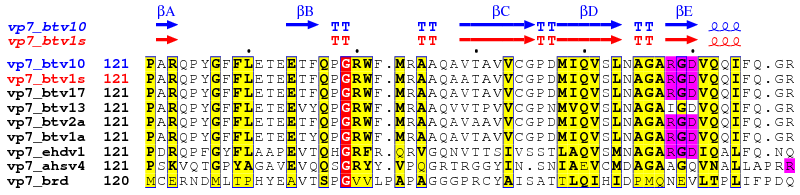
These examples above refer to a study made with the group of Prof. David STUART, Division of Structural Biology (Oxford) on viral proteins VP7 and VP3 in orbiviruses (19,20).
1. vp2_rota.inp (resulting PostScript, PNG)
vp2_rota.phd ! mail from the Predict Protein server on vp2 rotavirus
* A none ! shows predicted sec. str. elements and accessibility on the top of each block
.
.7 E ! physico-chemical boxing
6 81 ! layout
@pp ! extracts all infos from the Predict Protein file
@noname ! no names for sec. structures
.
2-6 ! sequences to be displayed
.
2. vp7_adv.inp (resulting PostScript, PNG)
vp7.aln vp7_btv10.pdb ! aligned sequences (from CLUSTALW) and pdb file
vp7_btv10.dssp A ! secondary structures (from DSSP)
vp7_adv.ps M ! PostScript output
.7 .5 R ! similarity criteria
7 60 ! layout
U R 127 250 ! -> red triangles
S B 168-170 178-180 ! -> blue stars
X B 1-126 254-349 ! -> sec. structure information in blue
X G 127-253 ! -> sec. structure information in green
T R 1-2 ! -> names of btv sequences in red
@noname ! no names for sec. structure
%Alignment for protein VP7.
.
2 1 3-6 ! first group of sequences
7-8 ! second group of sequences
9 ! third group of sequences
.
3. vp3_sup.inp (resulting PostScript, PNG)
vp3.aln + ! The + enables extra input
vp31001.art_dssp ! DSSP file for btv-10 vp3
vp3_sup.ps M ! PostScript output
.7 .5 R
5 160 13 8 0 C L N
T R 1 ! title 1 sequence in red
Y R all ! numbering 1 sequence in red
@noname ! no names for sec. structure
%Alignment vp3 orbivirus and attempt against vp2 rotavirus
. ! end special characters
1-2 ! 1st group of sequences
9 10 ! 2nd group of sequences
11 ! 3rd group of sequences
. ! end
vp2_casper.aln 8 ! 2.THREADER alignment between btv vp3 and rota vp2
vp31001.art_dssp vp2_rota.phd ! DSSP file for btv and PHD file for rota
vp3_sup.ps
.7 R
5 160 16 -1 0 C L N
T R 1 ! title 1 sequence in red
Y R all ! numbering 1 sequence in red
T B 2 ! title 2 sequence in blue
Z B 2 ! numbering 2 sequence in blue
@noname ! no names for sec. structure
. ! end special characters
. ! all sequences in one group
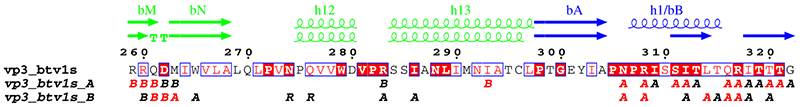
4. vp3_art.inp (resulting PostScript, PNG)
vp3.aln + ! CLUSTAL alignment for VP3 and option +
vp32001.art_dssp ! DSSP file for first monomer
vp3_art.ps
.7 R
6 129 8 0 0 C L
X G 1 ! domains
X D 1-1
X B 298-587
X R 699-856
@skip ! hide sequence
@noname ! no names for sec. structure
@ah3 bbC bbD bbE ah4 ah5 bbF bbG bbH ah6 bbI ah7 ah8 bbJ bbK ah9
@ah10 bbL ah11 bbM bbN ah12 ah13 bbA ah1/bB ah2 ah3 bbD ah4 ah5
@ah6 bbE bbF bbG ah7 bbH bbI bbJ/h8 ah9 ah10 ah11 bbK ah12 bbL
@bbQ ah14 ah15 ah16 ah17 bbP/h18 ah19 ah20 ah21 bbA bbB bbC ah1
@bbE bbF bbG ah2 ah3 bbH bbI ah4 bbJ bbK bbL ah5 bbM bbN bbQ ah22
@bbR bbS ah23 bbT ! new sec. structure labels
.
1 %10 %11 %12
.
vp3.aln vp3_contact.log ! same alignment and CNS output for contacts
vp31001.art_dssp ! DSSP file for second monomer
vp3_art.ps
.7 R
6 129 8 -1 0 C L ! vertical shift
X G 1 ! domains
X D 1-1
X B 298-587
X R 699-856
A S all ! no letters above sec. elements
R A all ! intermolecular contacts for VP3A
R B all ! intermolecular contacts for VP3B
@noname ! no names for sec. structure
%Secondary structures for vp3A and vp3B, article definition.
.
1 %10 %11 %12
.
5. vp7_exp.inp (resulting PostScript, PNG)
vp7.aln + ! CLUSTAL alignment on orbivirus
vp7_btv10.dssp ! btv10 secondary elements
vp7_exp.ps M
.
7 60 6 0 F N
X B 1 ! btv10 sec. elements in blue
@skip ! hide all sequences
@h ! first 310-helix is not labeled
%Alignment on VP7 with two secondary structure elements.
.
2 all ! btv10 in first then other sequences
.
vp7.aln ! same aligned sequences
vp7_btv1.dssp ! btv1 secondary elements
vp7_exp.ps M
.8 E ! homology criteria is %Equivalence
7 60 6 -1 F N ! vertical shift(-1) flashy colours(F) all sequences numbered(N)
X R 2 ! btv1 sec. elements in red
A S all ! remove sec. structure labels
T B 1 ! btv10 title in blue
T R 2 ! btv1 title in red
Q P 1-3 169-171 ! highlight RGD segment in pink
Q P 5-7 169-171
Q P 8 180-182
.
2 all ! btv10 is the first displayed sequence
.
| 1. |
Wootton, J. C. and Federhen, S. (1996) Analysis of compositionally biased regions in sequence databases. Meth. in Enzymol. 266, 554-571.
| | 2. |
Sigrist, C. J., de Castro, E., Cerutti, L., Cuche, B. A., Hulo, N., Bridge, A., Bougueleret, L. and Xenarios, I. (2013) New and continuing developments at PROSITE. Nucleic Acids Res. 41(Database issue), D344-347.
| | 3. |
Corpet, F. (1988) Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10881-10890.
| | 4. |
Bru, C., Courcelle, E., Carrère, S., Beausse, Y., Dalmar, S. and Kahn, D. (2005) The ProDom database of protein domain families: more emphasis on 3D. Nucleic Acids Res. 33(Database issue), D212-D215.
| | 5. |
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J., and Higgins, D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947-2948.
| | 6. |
Sievers, F., Wilm, A., Dineen, D. G., Gibson, T. J., Karplus, K., Li, W., Lopez, R., McWilliam, H., Remmert, M., Soding, J., Thompson, J. D. abd Higgins, D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Sys. Biol. 7, 539.
| | 7. |
Combet, C., Blanchet, C., Geourjon, C. and Deléage, G. (2000) NPS@: Network Protein Sequence Analysis. TIBS 25, 147-150.
| | 8. |
Pearson, W. R. (2014) BLAST and FASTA similarity searching for multiple sequence alignment. Methods Mol. Biol. 1079, 75-101.
| | 9. |
Gouy, M., Guindon, S. and Gascuel, O. (2010) SeaView version 4 : a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221-224.
| | 10. |
Berman, H. M., Battistuz, T., Bhat, T. N., Bluhm, W. F., Bourne, P. E., Burkhardt, K., Feng, Z., Gilliland, G. L., Iype, L., Jain, S., Fagan, P., Marvin, J., Padilla, D., Ravichandran, V., Schneider, B., Thanki, N., Weissig, H., Westbrook, J. D., and Zardecki, C. (2002) Acta Cryst. D58, 899-907.
| | 11. |
Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros P., Grosse-Kunstleve, R. W., Jiang J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice L. M., Simonson, T. and Warren, G. L. (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Cryst. D54, 905-921.
| | 12. |
Joosten, R. P., Te Beek, T. A. H., Krieger, E., Hekkelman, M. L., Hooft, R. W. W., Schneider, R., Sander, C. and Vriend, G. (2011) A series of PDB related databases for everyday needs. Nucleic Acids Res. 39(Database issue), D411-D419.
| | 13. |
Frishman, D. and Argos, P. (1995) Knowledge-based secondary structure assignment. Proteins 23, 566-579.
| | 14. |
Rost, B., Yachdav, G. and Liu, J. (2004) The PredictProtein server. Nucleic Acids Res. 32(Web Server issue), W321-W326.
| | 15. |
Kyte, J. and Doolittle, R. (1982) A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157, 105-132.
| | 16. |
Risler, J. L., Delorme, M. O., Delacroix, H. and Henaut, A. (1988) Amino acid substitutions in structurally related proteins. A pattern recognition approach. Determination of a new and efficient scoring matrix. J. Mol. Biol. 204, 1019-1029.
| | 17. |
Dayhoff, M. (1978) "Atlas of protein sequences and structure" National Biomedical Research Foundation. Washington, D.C., p. 345.
| | 18. |
Henikoff, J. G. and Henikoff, S. (1996) Blocks database and applications. Meth. in Enzym. 266, 88-105.
| | 19. |
Grimes, J. M., Burroughs, J. N., Gouet, P., Diprose, J. M., Malby, R., Zientara, S., Mertens, P. P. C. and Stuart, D. I. (1998) The atomic structure of the bluetongue virus core. Nature 395, 470-478.
| | 20. |
Gouet, P., Diprose, J. M., Grimes, J. M., Malby, R., Burroughs, J. N., Zientara, S., Stuart, D. I. and Mertens, P. P. C. (1999) The highly ordered double-stranded RNA genome of bluetongue virus revealed by crystallography. Cell 97, 481-490.
|
User guide last revision: January 18, 2016
|